")
")
Maladie de Pompe infantile chez un nourrisson porteur d'albinisme oculo-cutané au CHU-MEL de Cotonou avec confirmation biochimique et moléculaire.
Infantile Pompe disease in an infant with oculocutaneous albinism at the CHU-MEL in Cotonou, confirmed by biochemical and molecular testing.
ADJAGBA P1, HOUÉNASSI LC1, DIDAVI U1, YAKOUBOU A2, SORY R1, ALAO MJ2.
RESUME
La maladie de Pompe ou glycogénose de type II est une maladie génétique héréditaire à transmission autosomique récessive. La forme infantile classique est rare et rapidement mortelle. Il est rapporté ici, un cas de maladie de Pompe, chez un nourrisson de dix mois porteur d'albinisme oculo-cutané, avec une confirmation biochimique et moléculaire. Il avait été admis pour le bilan d’hypotrophie. L’examen clinique avait permis de retrouver un retard du développement psychomoteur et staturo pondéral, une hypotonie globale à prédominance axiale, une macroglossie et une hépatomégalie. L’électrocardiogramme montrait un aspect de pré excitation ventriculaire et une hypertrophie bi ventriculaire. L’échocardiographie retrouvait une hypertrophie biventriculaire importante, avec dysfonction systolique sévère. La confirmation diagnostique a été faite par le séquençage génétique révélant la présence d’un variant de séquence de l’exon 18 du gène GAA : c. 2560C>T, p. (Arg854) à l’état homozygote. Les parents étaient hétérozygotes pour ce variant. Le dosage de l’activité enzymatique a révélé un déficit en alpha-glucosidase acide (ou maltase acide) en faveur du diagnostic de la glycogénose de type II chez le propositus. Le traitement spécifique par l’enzymothérapie substitutive n’a pu être initié. L’évolution a été marquée par le décès du nourrisson une vingtaine de jours après la première consultation.
MOTS CLES
Maladie de Pompe, cardiomyopathie hypertrophique, séquençage, mutation génétique.
SUMMARY
Pompe disease, or type II glycogen storage disease, is an autosomal recessive genetic disorder. The classic infantile form is rare and rapidly fatal. We report here a case of Pompe disease in a 10-month-old infant with oculocutaneous albinism, confirmed by biochemical and molecular testing. He was admitted for evaluation of hypotrophy. Clinical examination revealed psychomotor and growth retardation, generalized hypotonia with axial predominance, macroglossia, and hepatomegaly. The electrocardiogram showed signs of ventricular pre-excitation and biventricular hypertrophy. Echocardiography revealed significant bi-ventricular hypertrophy with severe systolic dysfunction. The diagnosis was confirmed by genetic sequencing revealing the presence of a sequence variant in exon 18 of the GAA gene: c. 2560C>T, p. (Arg854) in a homozygous state. The parents were heterozygous for this variant. Enzyme activity testing revealed a deficiency in acid alpha-glucosidase (or acid maltase), supporting the diagnosis of type II glycogen storage disease in the proband. Specific treatment with enzyme replacement therapy could not be initiated. The infant died approximately 20 days after the initial consultation.
KEY WORDS
Infantile Pompe disease, hypertrophic cardiomyopathy, sequencing, genetic mutation.
1 Service de cardiologie adulte et pédiatrique, Centre Hospitalier Universitaire Mère Enfant Lagune, Cotonou, Bénin
2- Service de Pédiatrie et Génétique Médicale, Centre Hospitalier Universitaire Mère Enfant Lagune, Cotonou, Bénin
Adresse pour correspondance
ADJAGBA Philippe,
10 BP 259, Cotonou, Bénin.
Email : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
INTRODUCTION:
La maladie de Pompe ou glycogénose de type II est une maladie génétique héréditaire à transmission autosomique récessive [1]. Elle fut décrite pour la première fois en 1932 par un médecin néerlandais Johannes Cassianus Pompe, après l’autopsie d'une fillette de 7 mois chez qui on avait retrouvé une hypertrophie myocardique idiopathique et une faiblesse musculaire généralisée. Les explorations biologiques avaient permis de retrouver la présence d’un stockage massif de glycogène dans pratiquement tous les tissus [2]. Le mécanisme de la pathologie a été mieux élucidé dans les décennies qui ont suivies grâce à la compréhension du métabolisme du glycogène. En effet, en 1963 le biochimiste belge Henri-Géry Hers réalise que l'enzyme dégradant le glycogène (l’acide alpha-glucosidase) normalement retrouvé dans le lysosome est absent dans la maladie de Pompe [3]. Il s’agit donc d’une anomalie de stockage du glycogène lysosomal causé par un déficit en α-glucosidase acide (GAA) dû à des variations de séquence pathogène dans le gène GAA correspondant [1, 4]. La corrélation entre les génotypes et les phénotypes est stricte, en ce sens que les patients présentant le phénotype le plus sévère, la maladie de Pompe infantile classique, présentent deux mutations pathogènes, une dans chaque allèle GAA, qui empêchent la formation de GAA ou oblitèrent totalement sa fonction. Tous les patients présentant des phénotypes moins progressifs présentent au moins une variation de séquence qui permet une synthèse normale ou de faible niveau de GAA [5].
Au plan épidémiologique, il est difficile de déterminer avec précision l’incidence de la maladie de pompe du fait de l’existence de formes tardives [6]. Elle est estimée entre 1/9.000 et 1/40.000 [7]. On distingue généralement deux types en fonction du délai d'apparition des symptômes : la forme infantile (Infantile-Onset Pompe Disease pour IOPD), qui se manifeste avant 12 mois et la forme tardive (late-onset Pompe disease pour LOPD) caractérisée par un délai d’apparition supérieur à 12 mois [8]. La forme infantile est plus sévère avec un pronostic sombre [1]. Elle est plus rarement retrouvée avec une prévalence mondiale estimée à 2 cas pour 100.000 naissances [4]. Sa fréquence varie en fonction des groupes ethniques. Elle est plus fréquente chez les Africains. On l’estime à 1/138.000 dans la population caucasienne et 1/31.000 chez les personnes d'origine africaine [9]. Quelques cas cliniques ont été rapportés en Afrique [6, 7, 9- 11]. Des cas de diagnostic anténatal ont également été rapportés [4]. Le pronostic est sombre marqué par le décès avant le premier anniversaire [6,12]. L'albinisme oculo-cutané (AOC) est un groupe de maladies génétiques héréditaires caractérisées par une déficience de la production de mélanine, le pigment responsable de la couleur de la peau, des cheveux et des yeux. L’association de l’AOC avec la maladie de Pompe n’a pas été rapportée dans la littérature et semble plutôt fortuite. Il est rapporté ici un cas de maladie de Pompe chez un nourrisson de dix mois porteur d’une AOC.
OBSERVATION
Il s’agissait d’un nourrisson de 10 mois, de sexe féminin, adressé en consultation de cardiologie pédiatrique pour l’exploration d’une hypotrophie.
La patiente était la deuxième fille d’une fratrie utérine de deux enfants, dont l’aîné de sexe masculin, serait en bonne santé apparente. Il n’y avait pas de consanguinité, ni d’antécédents de cardiopathie familiale connue.
Le début de la symptomatologie remonterait à l’âge de trois mois, marqué par les symptômes suivants :
- une hypotonie d’aggravation progressive avec une gestualité pauvre,
- un cri faible quasi inaudible,
- une mauvaise prise pondérale,
- et une absence d’acquisition la tenue de la tête.
Une dyspnée apparue à l’âge de huit mois.
Elle pesait 6,130 kg pour une taille de 68 cm. Les rapports anthropométriques selon les courbes de l’OMS retrouvaient :
- une insuffisance pondérale modérée (rapport poids pour âge en dessous de -2 Zscore),
- un risque de retard statural (rapport taille pour âge en dessous de moins -1 Zscore),
- et une émaciation modérée (rapport poids pour taille en dessous de -2 Zscore).
L’examen retrouvait un état général conservé. Il y a avait un albinisme oculo-cutané (figure 1-A) et un visage peu expressif en plus d’une hypotonie massive avec un aspect de « Floppy baby» (Figure 1-A).
Au plan cardiovasculaire, la fréquence cardiaque était de 140 battements/min, la saturation en oxygène était de 96% en air ambiant. Les bruits du coeur étaient réguliers sans souffle. Il n’y avait pas de signes de défaillance cardiaque. On retrouvait par ailleurs une hépatomégalie et une macroglossie (figure 1-B).
Une maladie neuromusculaire a été évoquée devant l’hypotonie massive. Les Créatines PhospoKinases (CPK) étaient à 1435 UI/l.
Il y avait une cardiomégalie (figure 2) avec un Index Cardio Thoracique (ICT) à 0,76, sans présence de troubles ventilatoires.
L’électrocardiogramme (figure 3) retrouvait :
- un rythme sinusal régulier avec une fréquence cardiaque à 136 bpm,
- un espace PR court à 0,06s, un aspect de pré excitation ventriculaire avec une onde delta (Wolf Parkinson White),
- une hypertrophie biventriculaire et des ondes T négative de V3 à V6, en D1-aVL et en D2-D3-aVF.
A l’échocardiographie Doppler (figure 4) on retrouvait une hypertrophie biventriculaire importante, plus marqué sur le ventricule gauche (septum interventriculaire en diastole de 10 mm, paroi postérieure en diastole de 10mm), une altération de la fonction systolique ventriculaire gauche (FEVG mesuré en Simpson bi-plan à 30%). Les cavités cardiaques gauches étaient dilatées.
Après les premières explorations biologiques et électro-morphologiques, une maladie de Pompe a été suspectée avec la mesure de l’activité enzymatique de l’alpha-glucosidase acide qui était déficitaire (Service de Biochimie – UF de Biochimie métabolique de Hôpital Necker-Enfants malades, APHP). La recherche de mutation dans le gène GAA a été faite, avec la découverte d’un variant homozygote dans l’exon 18 [c. 2560C>T, p. (Arg854*)] chez la patiente d’une part, ainsi que la présence de la même mutation hétérozygote chez les deux parents d’autre part (Service de Médecine Génomique des Maladies de Système et d’Organe des Hôpitaux Universitaires, Paris Centre) confirmant le diagnostic de la maladie de Pompe.
Le traitement institué était fait d’un diurétique et d’un inhibiteur de l’enzyme de conversion par voie orale. Le traitement enzymatique de substitution indiqué dans ce cas, étant indisponible au Bénin et hors de prix pour les parents, n’a pu être démarré.
L'évolution a été rapidement défavorable marquée par l’installation des signes de défaillance cardiaque, suivis du décès du nourrisson à l’âge de onze mois, dans un tableau de détresse respiratoire.
Les parents du propositus ont été informés des différents résultats biochimique et moléculaire. Les différents entretiens avec eux ont porté non seulement sur la compréhension de la maladie découverte, mais également sur ces implications pour la vie de couple, notamment le risque de récurrence qui est de un sur quatre à chaque grossesse.
La possibilité d’un diagnostic anténatal précoce, par la mesure de l’activité enzymatique et surtout, par la recherche de la mutation dans le gène GAA chez le foetus a été évoquée avec les parents après amniocentèse à 16 semaines d’aménorrhée (SA).
DISCUSSION
Plusieurs intérêts peuvent être notés dans ce cas clinique de double portage d'affections génétiques potentiellement graves et mortelles :
- l'une dans l'immédiat (la maladie et Pompe),
- et l'autre (l’albinisme oculo-cutané) à long termes avec ses complications dermatologiques et oculaires.
Ce double portage d'affections génétiques n’est pas décrit dans la littérature et semble être fortuit.
La maladie de Pompe infantile se manifeste au cours des premiers mois de vie [8]. Chez cette patiente les symptômes ont débuté à l’âge de trois mois. Les enfants sont le plus souvent vus en consultation pour une insuffisance respiratoire ou des signes d’insuffisance cardiaque. L'hypotonie généralisée est un signe quasi constant avec la position en grenouille [9]. On retrouve une faiblesse musculaire proximale des membres, une hypotonie axiale avec incapacité de maintien de la tête. Le développement moteur est retardé et les étapes majeures du développement, telles que la capacité à se retourner, à s'asseoir ou à se lever, ne sont souvent pas atteintes [6].
Sur le plan digestif l’alimentation notamment la succion est rendue difficile par la faiblesse des muscles oropharyngés, ce qui donne lieu à un retard staturo pondéral. On observe une macroglossie et une hépatomégalie. Le dysfonctionnement progressif du diaphragme et des muscles respiratoires accessoires est responsable d’une insuffisance respiratoire. Les infections respiratoires sont récurrentes généralement dues à un réflexe de toux altéré et à une désobstruction inefficace des voies aériennes [9, 13]. La patiente rapportée ici n’a pas présenté de signes de défaillance respiratoire pouvant témoigner du dysfonctionnement des muscles respiratoires. Il n’y avait pas sur la radiographie pulmonaire, des troubles ventilatoires confirmant ainsi le caractère exceptionnel de l’absence d’atteinte respiratoire chez cette patiente.
L'atteinte cardiaque est fréquente et la majorité des nourrissons présentent une cardiomyopathie hypertrophique avant l'âge de 6 mois, avec une progression vers l'insuffisance cardiaque [14, 15]. L'échocardiographie est précieuse pour évaluer la structure et la fonction ventriculaires. L'hypertrophie, porte principalement sur la paroi postérieure du ventricule gauche et le septum inter ventriculaire, et peut être également biventriculaire. Elle est généralement observée chez les nourrissons au moment du diagnostic, comme ce fut le cas dans cette observation, mais peut également être identifiée avant la naissance [4, 8].
L’approche diagnostique de la maladie de Pompe infantile doit être syndromique. En effet la maladie de Pompe avec atteinte cardiaque, doit être alors considérée comme une cardiopathie syndromique. L’atteinte cardiaque est associée à une ou plusieurs malformations extracardiaques aisément reconnaissables (hypotrophie, hypotonie généralisée, macroglossie, hépatomégalie, infection respiratoire récurrentes) qui orientent vers le diagnostic. Vu sous cet angle, la maladie de Pompe avec atteinte cardiaque représentait 3,2% des cardiopathies syndromiques dans une précédente série publiée au Bénin [11]. Toutefois, il existe néanmoins des cas de maladie de pompe infantiles sans atteinte cardiaque chez des patients plus un âgé au moment des premiers symptômes et du diagnostic [16].
Les caractéristiques électrocardiographiques typiques de la maladie de Pompe comprennent un intervalle PR raccourci, un hyper voltage prononcé des complexes QRS et des anomalies de la repolarisation ; elles étaient toutes présentes chez la patiente. Des arythmies peuvent être présentes, notamment des tachyarythmies supraventriculaires [8, 6].
Le diagnostic de la maladie de Pompe est clairement établi en démontrant un déficit enzymatique de l'alpha-glucosidase acide et en identifiant les mutations responsables de la maladie dans le gène GAA [8].
La démonstration d'un déficit enzymatique de la GAA reste un critère de référence et les tests enzymologiques sont les plus utilisés pour le diagnostic de la maladie de pompe [17]. Il existe des tests biochimiques mesurant l'activité de la GAA dans des fibroblastes cutanés en culture, des biopsies musculaires ou des échantillons sanguins à l'aide de gouttes de sang total séché sur papier filtre, de lymphocytes purifiés et de leucocytes mixtes. La confirmation du déficit enzymatique chez la patiente présentée dans cette observation a été faite sur des tâches de sang sur papier buvard.
La maladie de Pompe est une maladie autosomique récessive, causée par une variante pathogène dans les deux copies du gène GAA. Le gène GAA est localisé sur le bras long du chromosome 17 (17q25.2 q25.3) et se compose de 20 exons [18]. La recherche de l’anomalie génétique est une étape importante dans la démarche diagnostique devant une maladie de Pompe. La recherche de la mutation a permis de retrouver la présence d’un variant homozygote de séquence de l’exon 18 du gène GAA : c. 2560C>T, p. (Arg854*) chez le nourrisson. La répartition des mutations du gène GAA varie selon l'origine ethnique. La mutation c.2560C>T (exon 18) retrouvée est fréquente dans la population afro-américaine, alors que la mutation la plus courante chez la population caucasienne est la variante intronique c.-32-13T>G [19]. Cette mutation c.2560C>T (exon 18) serait responsable de formes plus sévères de la maladie de Pompe.
La confirmation génétique ainsi que la mise en évidence du déficit de l’activité enzymatique, ne sont pas disponibles au Bénin. Le plateau technique au Bénin ne permettant pas la réalisation des tests de confirmation diagnostic sur place, les prélèvements sont envoyés à l’étranger pour la confirmation du diagnostic. C’est tout l’intérêt de la coopération multidisciplinaire entre les pays du Sud et ceux du Nord qui disposent bien souvent des infrastructures appropriées.
Pendant longtemps, les interventions thérapeutiques pour la maladie de Pompe se limitaient aux soins palliatifs. Le traitement enzymatique substitutif (TES) par GAA humaine recombinante (Myozyme®) a été approuvé dans l'Union européenne en 2006 et aux États-Unis (Lumizyme®) en 2010 [16, 12]. Récemment en 2022, une nouvelle enzymothérapie substitutive à base d’avalglucosidase alpha (Nexviadyme®) a été mise sur le marché pour le traitement au long cours des formes tardives et infantiles de la maladie de Pompe. L’enzymothérapie substitutive tend à améliorer le pronostic lorsqu’elle est administrée au début des manifestations cliniques et réduit l’atteinte cardiaque [6]. Cependant ce traitement reste très onéreux. Le coût hospitalier et non
hospitalier associé à la forme infantile de la maladie de Pompe chez les patients traités avec Myozyme® a été estimé à 232 117 € en 2022 [20]. Le nourrisson présenté ici n’a pu bénéficier de ce traitement car il est indisponible au Bénin et hors de prix pour la famille.
L’évolution a été défavorable en quelques jours. L’évolution spontanée de la maladie de Pompe est rapidement défavorable, marquée par la survenue d’une dysfonction diastolique puis systolique du ventricule gauche avec installation d’une insuffisance cardiaque, une majoration des signes respiratoires évoluant vers une détresse respiratoire avec nécessité d’une assistance respiratoire. Seul un faible pourcentage de patients non traités survit au-delà d'un an, la principale cause de décès étant l'insuffisance cardiaque et respiratoire [1, 6].
CONCLUSION
La maladie de Pompe ou anomalie de stockage du glycogène est une maladie rare et grave due à un déficit en alpha glucosidase. La forme infantile classique est plus fréquente chez les sujets africains et est associée à une mortalité importante. Le diagnostic doit être évoqué devant une hypotonie et une cardiomyopathie hypertrophique chez un nourrisson ou un nouveau-né. La confirmation du déficit enzymatique et la mise en évidence de la mutation génétique sont un véritable challenge dans les pays à revenu limité. Un conseil génétique avant la procréation de même que la possibilité d’un diagnostic anténatal chez porteurs hétérozygotes pourraient réduire son incidence.
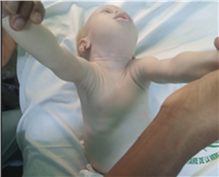
Figure 1-A : Aspect de Floppy baby
chez un enfant de 10 mois, porteur de
maladie de la Pompe infantile et d’un
albinisme oculo-cutané
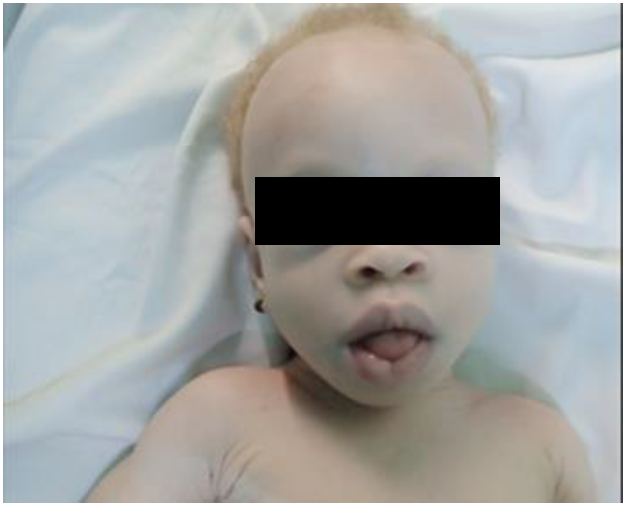
Figure 1-B : Macroglossie chez un
enfant de 10 mois, porteur de maladie
de la Pompe infantile et d’un
albinisme oculo-cutané
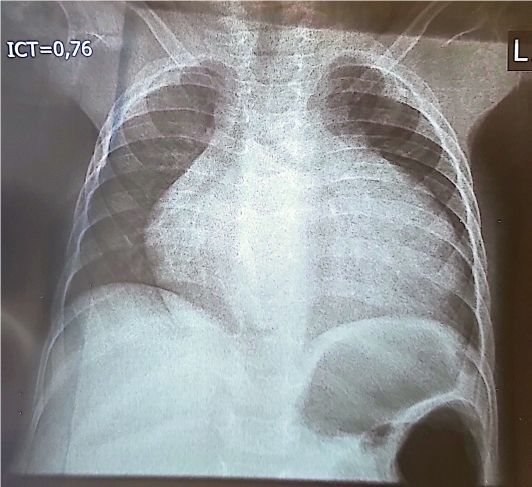
Figure 2 : Radiographie pulmonaire de face, chez un enfant de 10 mois,
porteur de maladie de la Pompe infantile et d’un albinisme
oculo-cutané, montrant une cardiomégalie avec ICT à 0,76
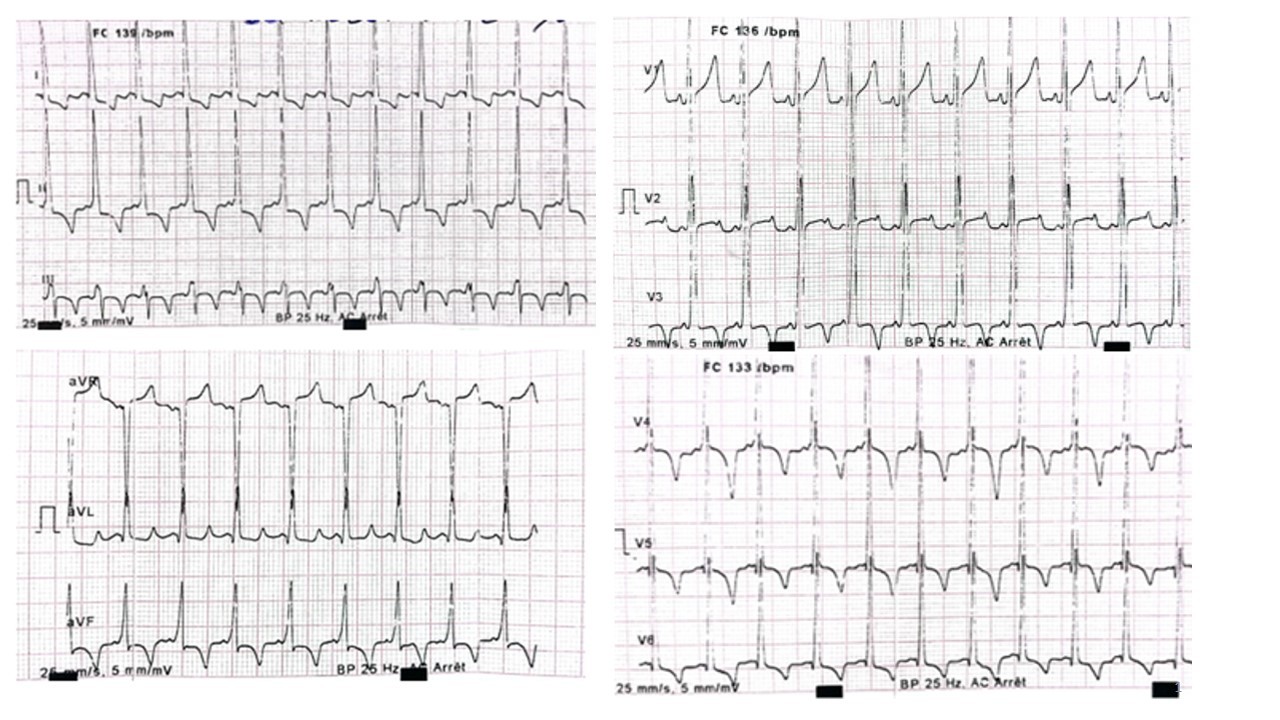
Figure 3 : ECG 12 dérivations, réalisé chez un enfant de 10 mois, porteur de maladie de la Pompe infantile et d’un albinisme oculo-cutané, à 25 mm/s et à 5mm/mV, retrouve un rythme sinusal régulier avec fréquence cardiaque à 136 bpm, un PR court à 0,07s, un aspect de pré excitation ventriculaire avec onde delta (Wolf Parkinson White), une hypertrophie bi ventriculaire et des ondes T négative de V3 à V6 et D1-aVL et D2-D3-aVF.
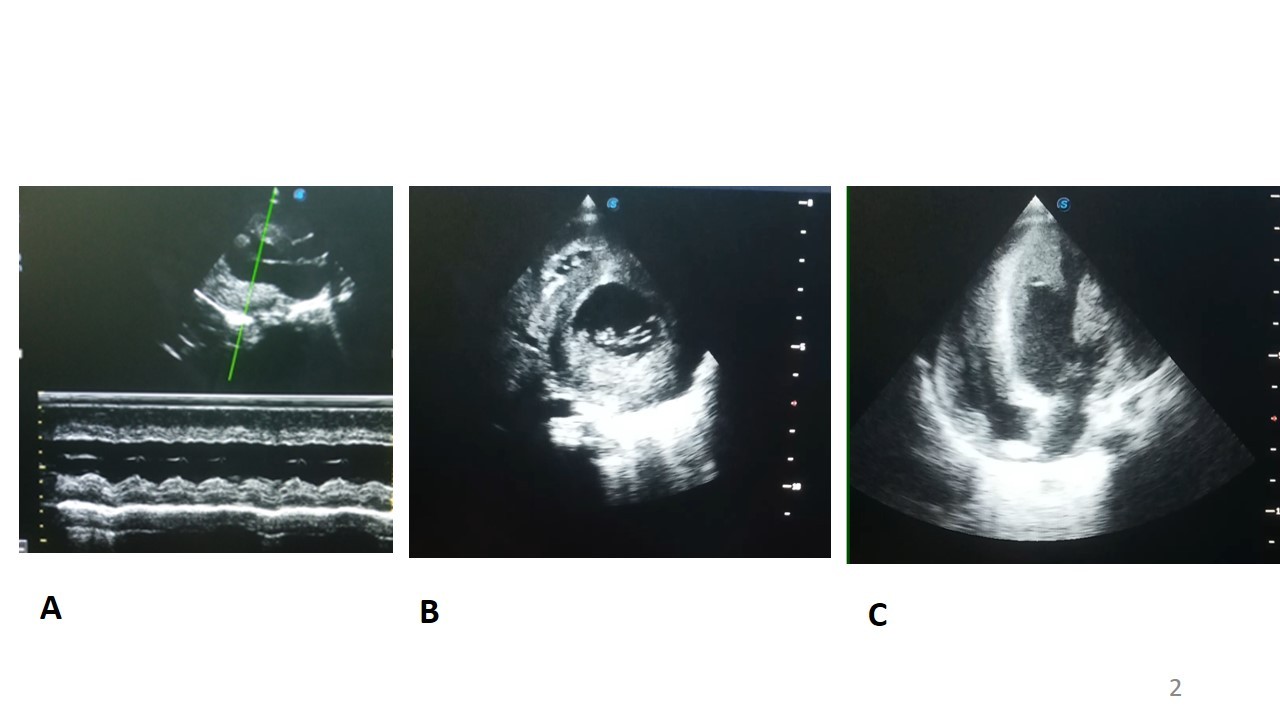
Figure 4 : Echocardiographie Doppler chez un enfant de 10 mois, porteur de maladie de la
Pompe infantile et d’un albinisme oculo-cutané.
A : mode TM en parasternal grand axe, montrant une hypertrophie concentrique du ventricule gauche avec septum intreventriculaire et paraoi postérieure mesurée en diastole à 10 mm
B : mode 2D en parasternal court axe, montrant une hypertrophie concentrique du ventricule gauche
C : mode 2D en apical 4 cavités montarnt une hypertrophie bi ventriculaire importante
|
REFERENCES |
|
1. Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics. 2018;15(4):928-942. 2. Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R et al. Diagnosis and Glucosidase deficiency in generalized glycogenstorage disease (Pompe's disease). Management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689-701. 3. Hers HG. Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe's disease). Biochem J. 1963;86(1):6-11. 4. Xi H, Li X, Ma L, Yin X, Yang P, Zhang L. Infantile Pompe disease with intrauterine onset: a case report and literature review. Ital J Pediatr. 2022;48(1):187. 5. Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C: Semin Med Genet 2012;160(1):59–68 6. Mpori JM, Tangni C, Allognon MC, Mfoumou A, Bivigou Mboumba B, Ayo Bivigou E, et al. Maladie de Pompe Infantile Classique de Découverte Fortuite : A Propos d'un Cas. Sciences de la santé. Dis. [Internet]. 23 décembre 2023 [cité le 17 mai 2025]. Disponible sur : http://www.hsd-fmsb.org/index.php/hsd/article/view/5157 7. Adadi N, Sahli M, Egéa G, Ratbi I, Taoudi M, Zniber L, et al. Post-mortem diagnosis of Pompe disease by exome sequencing in a Moroccan family: a case report. J Med Case Rep. 2018;12(1):322. 8. Tarnopolsky M, Katzberg H, Petrof B, Sirrs S, Sarnat HB, Myers K, et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. Canadian Journal of Neurological Sciences. 2016 ; 43(4), 472-485. 9. Sifi Y, Medjroubi M, Froissart R, Taghane N, Sifi K, Benhabiles A, et al. Clinical Analysis of Algerian Patients with Pompe Disease. J Neurodegener Dis. 2017;2017:9427269. 10. Folayan OS, Agaja OT, Adebayo BE, Ogunkunle O, Omokhodion SI. A case series of infantile Pompe disease. Progress in Pediatric Cardiology. 2022; 66:101538. 11. Adjagba P, Kétoh K, Bagnan L, Hounkponou M, Sonou A, Houénassi DM, et al. Les cardiopathies congénitales syndromiques au CNHU-HKM de Cotonou, Bénin. Revue Tunisienne de Cardiologie. 2020;16 (3) 169-176. 12. Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, Herbelin L, et al. Pompe disease: literature review and case series. Neurol Clin. 2014;32(3):751-76, ix. doi: 10.1016/j.ncl.2014.04.010. PMID: 25037089; PMCID: PMC4311397. 13. Winkel LPF, Hagemans MLC, Van Doorn PA, Loonen MCB, Hop WJC, Reuser AJJ, et al. The natural course of non-classic Pompe's disease; a review of 225 published cases. J Neurol. 2005 Aug;252(8):875-84. 14. Soliman OII, Van Der Beek NAME, Van Doorn PA, Vletter WB, Nemes A, Van Dalen B M, et al. Atteinte cardiaque chez l'adulte atteint de la maladie de Pompe. Journal of Internal Medicine. 2008; 264(4) : 333–339. 15. Jegadeeswari A, Amuthan V, Janarthanan RA, Murugan S, Balasubramanian S. Two cases of Pompe's disease: case report and review of literature. Indian Heart J. 2012 Mar-Apr;64(2):214-6. 16. Byrne BJ, Kishnani, PS, Case LE, Merlini L, Müller Felber W, Prasad S, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103(1):1-11; Erratum in: Mol Genet Metab. 2011;104(3):424. 17. Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J. Methods of diagnosis of patients with Pompe disease: Data from the Pompe Registry. Mol Genet Metab. 2014;113(1-2):84-91. doi: 10.1016/j.ymgme.2014.07.014. Epub 2014 Jul 16. PMID: 25085280. 18. Taverna S, Cammarata G, Colomba P, Sciarrino S, Zizzo C et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020;12(15):15856-15874. 19. Taverna S, Cammarata G, Colomba P, Sciarrino S, Zizzo C, Francofonte D, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020;12(15):15856-15874. 20. Le Bras A, Nze Ossima A, De Lonlay P, Attarian S, Durand-Zaleski I, Laforêt P. Analyse des dépenses de santé et des parcours de soins des patients atteints de la maladie de Pompe recevant du Myozyme : une étude observationnelle basée sur les données du système national des données de santé (SNDS). Journal of Epidemiology and Population Health. 2025; 73S1 : 202882 |