")
")
Cas Clinique . Maladie de Takayasu révélée par une insuffisance aortique massive.
Takayasu's arteritis revealed by massive aortic regurgitation.
NIAMKEY T1, EHLAN F1, ANGORAN I1, ANZOUAN-KACOU JB1, C KONIN2
RESUME
La maladie de Takayasu (MT) est une artérite inflammatoire chronique rare touchant les gros vaisseaux. Elle est très peu décrite en Afrique subsaharienne, où le diagnostic est souvent tardif au stade de complications. Son association à une valvulopathie est inhabituelle et est souvent une circonstance de découverte. Nous rapportons le cas d’une patiente dont la maladie a été révélée par une insuffisance aortique massive après un long parcours de soins.
MOTS CLES
Maladie de Takayasu, insuffisance aortique, Afrique subsaharienne
SUMMARY
Takayasu's disease is a rare chronic inflammatory arteritis affecting large vessels. It is very rarely described in sub-saharan Africa, where the diagnosis is often late in complications. Its association with valvular heart disease is unusual and often a circumstance of discovery. We report the case of a patient whose disease was revealed by a massive aortic regurgitation after a long course of care.
KEY WORDS
Takayasu disease, massive aortic regurgitation, sub-Saharan Africa.
1.Service des Explorations Externes de l’Institut de Cardiologie d’Abidjan (Côte d’Ivoire)
2.Service des Soins Intensifs de l’Institut de Cardiologie d’Abidjan (Côte d’Ivoire)
Adresse pour correspondance
Dr NIAMKEY Thierry
Service des Explorations Externes de l’Institut de Cardiologie d’Abidjan (Côte d’Ivoire)
Tel: 0022547440404
Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
INTRODUCTION
La maladie de Takayasu (MT) est une artérite inflammatoire chronique de la femme jeune touchant essentiellement les gros vaisseaux [1]. Elle est réputée rare en particulier en Afrique subsaharienne, où le diagnostic est souvent tardif et fait à distance de la phase systémique [2 ; 3]. Son association à une valvulopathie est inhabituelle et est souvent dans notre contexte une circonstance de découverte de la maladie. Nous en rapportons un cas.
OBSERVATION
Melle T.S âgée de 31 ans sans antécédents cardiovasculaires connus est vue en consultation en septembre 2014 pour une dyspnée d’effort et des précordialgies d’effort évoluant depuis près de 8 mois.
A l’examen clinique, elle présentait un bon état général, sans syndrome infectieux avec un poids de 55 kg pour une taille de 1.55 m. Les pouls périphériques étaient perçus de façon symétriques, amples et bondissants, avec un élargissement de la différentielle de la pression artérielle à 150/40 mm Hg, constatée aux 2 bras. L’auscultation cardiaque retrouvait des bruits du cœur réguliers avec un souffle holodiastolique 3/6 doux, aspiratif maximum le long du bord gauche du sternum et bien perçu en antéflexion. On ne notait pas de signes périphériques de défaillance cardiaque. Le diagnostic d’insuffisance aortique importante est évoqué, confirmé par une échocardiographie transthoracique et une étiologie rhumatismale est suspectée à cette période. Un remplacement valvulaire est proposé mais non réalisé en raison de difficultés financières. Après trois années de suivi médical irrégulier, la patiente signale des paresthésies du membre supérieur gauche. L’examen clinique notait alors en plus des signes périphériques d’insuffisance aortique, une abolition des pouls radial, ulnaire et brachial du membre supérieur gauche, une pression artérielle non perceptible au bras gauche et un souffle systolique carotidien gauche.
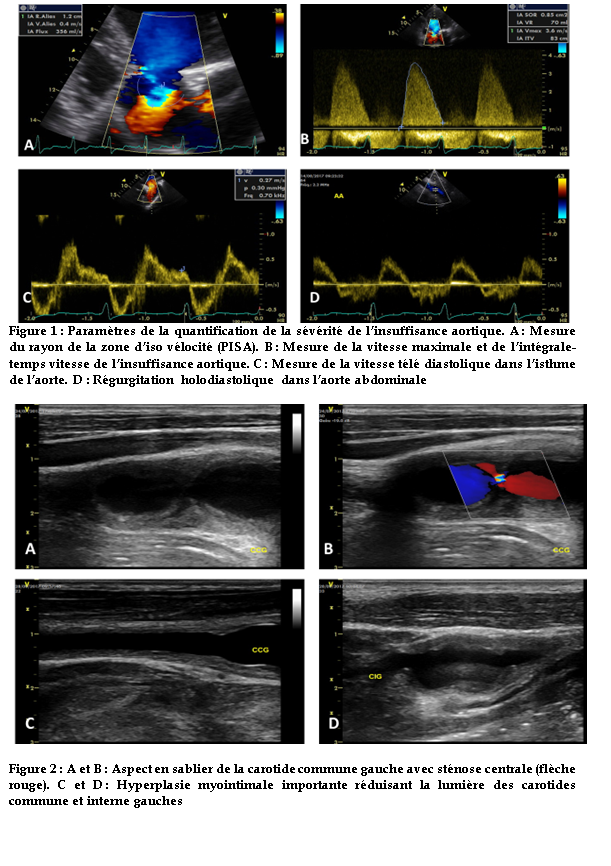
L’échocardiographie transthoracique de réévaluation objectivait une insuffisance aortique importante (VC = 8 mm ; Rayon PISA = 12 mm ; Surface de l’Orifice Régurgitant (SOR) = 85 mm² ;Volume de régurgitation (VR) = 70 ml/battement ; vitesse télédiastolique dans l’isthme = 27 cm/s ; reflux holodiastolique dans l’aorte abdominale).
Cette régurgitation aortique était de type 3 avec un épaississement et une restriction valvulaire importante responsable d’un diastasis central. L’aorte ascendante était de taille normale.
Il y était associé une insuffisance mitrale minime de même mécanisme, une dilatation importante du ventricule gauche (42 mm/m²) avec une altération moyenne de la fonction systolique (FEVG = 30-35%) et une dilatation importante de l’oreillette gauche (VOG = 60 ml/m²). (Figure 1)
L’écho doppler des troncs supra-ortiques (figure 2) mettait en évidence un double anévrysme non thrombosé séparé par une striction de la carotide commune gauche réalisant un aspect en sablier. On notait une hyperplasie myointimale circonférentielle, régulière et bilatérale à prédominance gauche avec une lumière résiduelle de 2.5 mm intéressant les carotides communes et internes sans retentissement hémodynamique sur le lit d’aval.
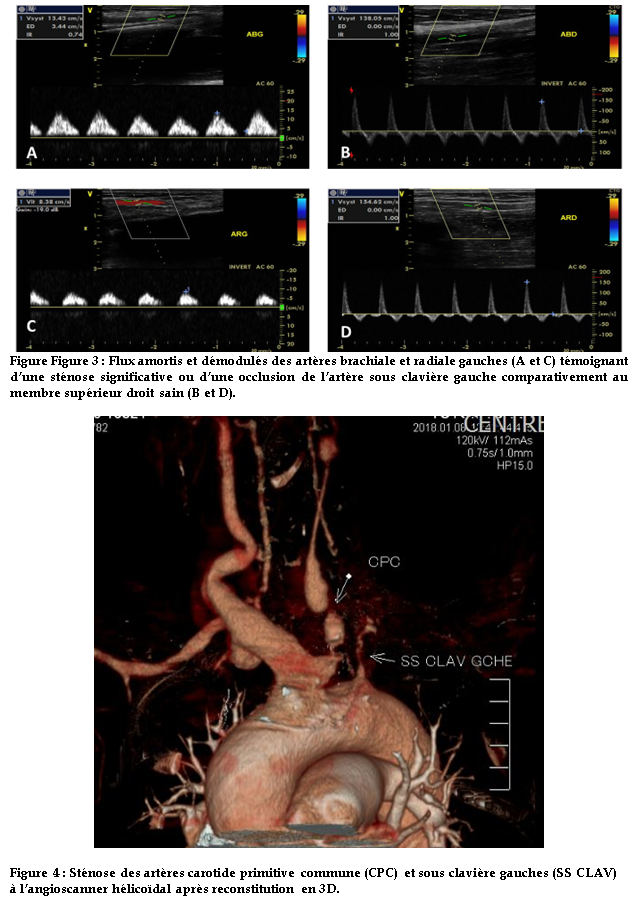
Il y avait par ailleurs à l’écho doppler des membres supérieurs, une sténose significative de l’artère sous clavière gauche à son origine responsable d’un amortissement et d’une démodulation des flux d’aval (figure 3).
L’angioscanner hélicoïdal centré sur les structures cervicales après reconstructions MPR et 3D confirme les sténoses des artères carotides et de la sous-clavière gauche (figure 4).
Les examens biologiques retrouvent un syndrome inflammatoire avec une CRP à 40 mg/l et une anémie hypochrome microcytaire à 9 g/dl. L’enquête tuberculeuse réalisée précédemment était négative.
Le diagnostic de maladie de Takayasu au stade occlusif a été retenu devant la présence de 6critères sur 6de l’American College of Rheumatology : Age de début inférieur à 40 ans ; claudication vasculaire du membre supérieur gauche ; abolition du pouls brachial ; différence de pression artérielle supérieure à 10 mmHg, souffle sur l’artère carotidienne gauche et la sténose carotidienne gauche et sous clavière gauche.
Un traitement comprenant un inhibiteur d’enzyme de conversion, un diurétique de l’anse et d’une faible dose de bétabloquant a permis une stabilité hémodynamique. Un remplacement valvulaire aortique et une endartériectomie carotidienne gauche sont envisagés.
DISCUSSION
La maladie de Takayasu est une pathologie rare touchant 1,2 cas par million d’habitants par an en Europe, environ 2,6 cas par million d’habitants par an en Amérique du Nord [1]. Sa fréquence est plus importante en Asie du Sud-est, en Inde et en Amérique du Sud. Quelques séries hospitalières sont décrites dans les pays du Maghreb [4]. En Afrique subsaharienne, les publications sur cette pathologie sont rares et portent sur de très faibles échantillons [2, 3]. Cette situation pose le problème de dépistage des maladies rares dans un continent en proie à lutter contre les pathologies infectieuses telles que le SIDA, la tuberculose et le paludisme [5]. La priorité est accordée aux maladies posant de réels problèmes de santé publique.
La MT évolue en deux phases. La première phase, dite pré-occlusive ou systémique, est caractérisée par des signes généraux avec une fièvre, des arthralgies, des myalgies, des signes cutanés et par un syndrome inflammatoire biologique. Elle passe souvent inaperçue [1]. La fréquence de cette phase inflammatoire varie selon les séries publiées et les modes de recrutement. Cette phase n’est décrite que dans 32 à 33% des cas [6]. Dans notre contexte, cette phase est souvent source d’erreur voire d’égarement diagnostique en raison de la non spécificité des signes. Ces manifestations cliniques orientent le plus souvent vers des pathologies infectieuses en particulier la tuberculose qu’il convient de formellement éliminer puisque son association à la MT est décrite dans la littérature. C’est le cas de notre patiente dont le parcours de soins a été d’au moins cinq ans avant le diagnostic de MT. En raison de la fréquence des valvulopathies, le plus souvent post rhumatismales, le diagnostic peut être confondu avec une poussée rhumatismale.
La deuxième phase dite occlusive se traduit par la survenue de manifestations cliniques ischémiques [1]. La diminution ou l’abolition d’un pouls périphérique, ainsi que l’apparition progressive d’une claudication vasculaire sont les signes cliniques qui révèlent fréquemment la maladie [1, 6].C’est à ce stade tardif que le diagnostic est évoqué dans notre contexte comme ce fut le cas chez notre patiente. L’usage courant des critères diagnostiques de l’American College of Rheumatology(ACR) mis au point en 1990[7] pourrait réduire les erreurs diagnostiques. En effet, cette classification repose surtout sur des symptômes cliniques (5 sur 6 critères). De plus, la présence de trois critères sur six permet de poser le diagnostic de MT avec une sensibilité de 90.5% et une spécificité de 97.8%.Ainsi, un interrogatoire minutieux, un examen clinique aussi exhaustif que possible peuvent permettre de dépister la MT, car l’artériographie est un examen invasif et l’imagerie de coupe est encore peu accessible dans nos pays en voie de développement.
Par ailleurs, L’association d’une MT avec une insuffisance aortique est inhabituelle [8]. La MT partage quelques similitudes épidémiologiques avec les valvulopathies rhumatismales qui restent fréquentes en Afrique subsaharienne et touche les sujets jeunes pauvres surtout de sexe féminin avec un risque accru de complications cardiovasculaires [9, 10]. L’étiologie rhumatismale est souvent évoquée à tort comme chez notre patiente. La conséquence est un long parcours de soins et une évolution de la maladie vers des complications cardiaques sévères. Une insuffisance aortique chez une femme de moins de 40 ans doit faire rechercher outre une étiologie rhumatismale, une artérite inflammatoire. Ce dépistage précoce permettra de mettre rapidement en route le traitement de première intention qu’est la corticothérapie.
CONCLUSION
La MT est une artérite réputée rare en Afrique subsaharienne. Sa découverte est souvent tardive en raison des erreurs diagnostiques. L’usage systématique des critères de classification de l’ACR devrait aider à la dépister plus précocement afin d’éviter des complications telles qu’une insuffisance aortique importante.
REFERENCES
1.Hiratzka et al. 2010 Guidelines on Thoracic Aortic Disease. JACC 2010;55(14) : 27–129.
2.Yangni-AngateH, Ayegnon G, Meneas C et al. Les artérites du noir africain : une expérience chirurgicale ivoirienne. Ann. Afr. Chir. Thor. Cardiovasc. 2008;3(1):19-26
3.Konin KC, Adoh AM,Coulibaly I et al. La maladie de Takayasu chez le noir africain: aspects cliniques et radiologiques. Cardiologie tropicale 2002 ; 28(112) : 59-63
4.La maladie de Takayasu en Tunisie : Etude mono-centrique de 11 cas. La Tunisie Médicale 2012 ; 90 (12) : 867 – 72.
5.Côte d’Ivoire Institut National de la Statistique. Enquête démographique et de santé, côte d’ivoire 1998-1999 Abidjan: Institut National de la Statistique, 1998.
6.Mirault T, Emmerich J. Maladie de Takayasu : Comment la prendre en charge ? Presse Med. 2012; 41: 975 – 85.
7.Arend WP, Michel BA, Bloch DA et al.The American College of Rheumatology 1990: criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990. 33(8):1129-34.
8.Alali WM, Alahmari SA, Alhebaishi YS and al. Severe aortic regurgitation complicating Takayasu's arteritis.Saudi Med J. 2017; 38 (8); 863-67
9.Kingué S, Abdou Ba S, Balde D and al. The VALVAFRIC study: A registry of rheumatic heart disease in Western and Central Africa. Archives of Cardiovascular Disease 2016; 109, 321—29.
10.Zuhlke L, Engel M, Karthikeyan G and al. Characteristics, complications, and gaps in evidence-based interventions in rheumatic heart disease: the Global Rheumatic Heart Disease Registry (the REMEDY study). European Heart Journal 2015; 36, 1115–22.